|
|
如何解决取向优势?
Bio3DEM张凯等
原文链接:
https://mp.weixin.qq.com/s/Bb4ZKNWvhDE0YHXh_BQcPg
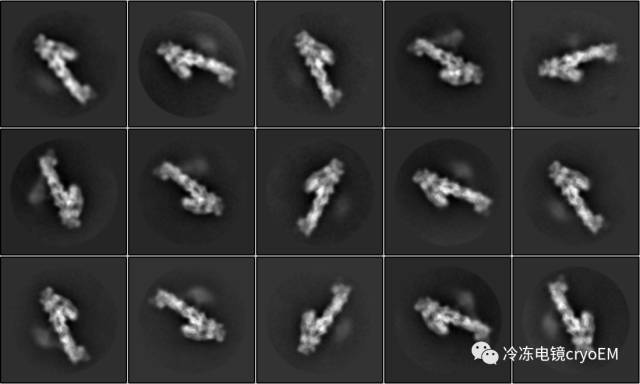
大哥你站起来,好不好?偏头痛啊,全都侧躺着!
如何解决取向优势?
冷冻电镜单颗粒分析从二维投影图获得三维密度图有一个基本前提:获得具有足够取向的全同颗粒。这个“足够取向”一般而言不需要全空间的投影图,只需要实验所获取的取向能够充分填充三维密度图傅里叶变换的系数即可。在最低的要求下,只需要绕着单轴旋转180度即可。然而,事实上在冷冻电镜的实验中,有大量情况只有一个或少数几个主导取向。如何解决取向优势是冷冻电镜实验和数据处理中遇到的最麻烦的问题之一。下面我们就简单总结一下可能的解决途径:
可能的解决方案:
1.样品自身性质的改变:
1.1 突变、截短或插入肽段:
重点考虑的对象为那些位于表面的带电氨基酸或暴露在外面的疏水肽段。以二型分子伴侣素为例,在顶部的盖子结构域有部分疏水氨基酸暴露,戒掉盖子的伴侣素取向会明显变好。
1.2 蛋白表面残基的修饰(如甲基化等)
1.3 交联试剂:
例如,0.01%-0.05%戊二醛制样前冰上处理5min左右;或使用较温和的BS3处理30min-1小时。
1.4 与co-factor、抗体等形成复合物
2.溶液性质改变:
2.1 缓冲液基本性质的改变:
盐浓度、pH值、缓冲试剂类型
2.2 添加去污剂:
不要认为只有膜蛋白需要去污剂,对于一些亲水蛋白,有可能会有局部的少数疏水氨基酸暴露,添加极少数的去污剂不仅可以使得蛋白溶液更容易形成冰层,而且有可能改变蛋白自身的表面性质,从而解决取向优势。去污剂必须为温和型,否则有可能严重影响蛋白结构。Triton X-100,NP-40,Tween-20,Digitonin等。
2.3 添加氨基酸(比如甘氨酸)或其它有机小分子:
2.4 增加冰层厚度:
很多蛋白分子不一定为近似圆形,有时候最长和最短的方向能差好几倍。由于冰层上下厚度的限制,当冰层和分子尺寸相当时,很多分子会优先“平躺”,也即Z-轴方向为分子较短的方向。经验表明,对这种情况增加冰层厚度可以有效增加取向,同时也保护了样品不被冰层表面张力损坏。在不影响目标蛋白前提下,可以考虑混入稳定的病毒或核糖体增厚冰层。
3.载网及处理:
3.1 尝试不同载网及同一种载网不同型号:
Quantifoil和C-flat Holy Carbon grid、微筛载网(Lacey carbon grid)、纯金载网(UltraAuFoil )(Russo andPassmore, 2014, 2016)
更多有关载网的信息可参照(此处非广告!):
http://www.quantifoil.com/index.php?name=products
3.2 额外支持膜:
例如纯冰或加额外的支持膜。额外的支持膜可以是无定形碳膜(amorphous carbon layer)或石墨烯(Graphene)或氧化石墨烯(Graphene Oxide)。氧化石墨烯改变取向最成功的例子可参看(Boland et al., 2017)。
3.3 辉光放电(glow discharge)和等离子清洗(plasma cleaning)条件。
例如可是尝试在不同气体环境:比如空气或在正戊胺(N-amylamine)(Nguyen et al., 2015)环境下做辉光放电,或氢气、氧气等环境做等离子清洗。不同气体解离形成不同电荷,比如H2解离带形成H+带正电,O2解离形成O2-带负电等,这样可以改变载网亲水处理之后的表面性质,从而改变与蛋白质的相互作用,最终使得被成功吸附的蛋白分子取向分布出现显著变化。
3.4 抗体亲和载网法:
具体可参见(Yu et al.,2016; Yu et al., 2014)。
3.5 Polylysine处理:
具体可参见(Lander et al., 2012; Zang et al., 2016)
4.数据收集和处理策略
4.1 暴力破解法:
此方法就是所谓的狂收数据策略。不过这个有一个前提,就是目标蛋白除了主要的取向,其它稀有取向概率分布并不为零;有些情况,例如一些环状结构,只有正面图像,没有侧视图,这种情况,完全无法使用狂收数据的方式解决。具体情况如何,必须在处理完2D分类之后再具体分析。
4.2 数据处理平衡取向:
当然这个是基于上条的暴力破解。简单说就是大量2D分类之后,把主要的优势取向混合,然后随机扔掉大部分颗粒。这个过程具体扔掉多少要看取向有多严重,以及自己有多少颗粒(百万级土豪随意)。具体操作,可以通过尝试法不同比例看那个结果最好,比方95%,90%,85%,80%,70%,60%,50%都试一下,只要有足够的计算资源,这是最有效的捷径。这个过程可以应用到3D,也可以迭代,也即等有了好的reference之后,取向也算的更加准确,这个时候把以前扔掉的颗粒再重新捡回来处理,再扔,再处理,直到满意为止。具体细节可参照dynactin(Urnavicius et al., 2015)和动力蛋白dynein(Zhang et al 2017)冷冻电镜数据处理,更多细节可email:kzhang@mrc-lmb.cam.ac.uk
4.3 倾转样品台:
一般而言,蛋白分子在平面内360度分布是完全均匀的,但是这没有意义,所有的取向都是旋转等价的,并不会贡献任何新的傅里叶信息。倾转可以大量增加有效取向,即使原来只有一个取向,也会转换为大量互不等价的取向。比如0度时在两极分布(就相当于地球的南北极),通过50度倾转样品台,分布变为整个北纬40度绕地球一圈(如果能绕赤道走一圈,就相当于完全没有取向优势,这等价于倾转90度,实际实验不可行)。倾转样品drift非常严重,要求grid的机械性质保持良好、不弯不折、无裂纹,样品自身分散要好,不要呈现大量团状聚集,冰层厚度尽量平整均一等等。
4.4 倾转数据后处理:
倾转样品两个关键因素就是drift和CTF。Drift可使用最新版的Motioncorr2中Patches功能,把Movie划分为若干小区域处理。CTF可以在做完Drift correction,挑完颗粒之后,有了坐标文件之后(.box或.star均可),用Gctf(Zhang, 2016)的local CTF功能做local refinement;也可以只算全局CTF,然后按样品台倾转参数+颗粒坐标计算欠焦量补偿值,然后把这个补偿值添加到全局CTF。
致谢:
Bio3DEM俞赞临(UCSF)、桂淼(清华大学)、吴惊香(北京大学)、李鲲鹏(普渡大学)等对本文撰写和校对等提出宝贵意见,特此致谢!
参考文献:
Boland, A., Martin,T.G., Zhang, Z., Yang, J., Bai, X.C., Chang, L., Scheres, S.H., and Barford, D.(2017). Cryo-EM structure of a metazoan separase-securin complex at near-atomicresolution. Nat Struct Mol Biol 24,414-418.
Lander, G.C., Estrin, E.,Matyskiela, M.E., Bashore, C., Nogales, E., and Martin, A. (2012). Completesubunit architecture of the proteasome regulatory particle. Nature 482, 186-191.
Nguyen, T.H., Galej, W.P., Bai,X.C., Savva, C.G., Newman, A.J., Scheres, S.H., and Nagai, K. (2015). Thearchitecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 523, 47-52.
Russo, C.J., and Passmore, L.A.(2014). Electron microscopy: Ultrastable gold substrates for electroncryomicroscopy. Science 346,1377-1380.
Russo, C.J., and Passmore, L.A.(2016). Ultrastable gold substrates: Properties of a support forhigh-resolution electron cryomicroscopy of biological specimens. J Struct Biol 193, 33-44.
Urnavicius, L.*, Zhang, K.*,Diamant, A.G., Motz, C., Schlager, M.A., Yu, M., Patel, N.A., Robinson, C.V.,and Carter, A.P. (2015). The structure of the dynactin complex and itsinteraction with dynein. Science 347,1441-1446.
K. Zhang*, H.E. Foster* et al. (2017).“Cryo-EM Reveals How Human Cytoplasmic Dynein IsAuto-inhibited and Activated.”Cell, 169, 1303–1314.http://www.cell.com/cell/fulltext/S0092-8674(17)30585-8
Yu, G., Li, K., Huang, P., Jiang,X., and Jiang, W. (2016). Antibody-Based Affinity Cryoelectron Microscopy at2.6-A Resolution. Structure 24,1984-1990.
Yu, G., Vago, F., Zhang, D.,Snyder, J.E., Yan, R., Zhang, C., Benjamin, C., Jiang, X., Kuhn, R.J., Serwer, P., et al. (2014). Single-stepantibody-based affinity cryo-electron microscopy for imaging and structuralanalysis of macromolecular assemblies. J Struct Biol 187, 1-9.
Zang, Y., Jin, M., Wang, H., Cui,Z., Kong, L., Liu, C., and Cong, Y. (2016). Staggered ATP binding mechanism ofeukaryotic chaperonin TRiC (CCT) revealed through high-resolution cryo-EM. NatStruct Mol Biol 23, 1083-1091.
Zhang, K. (2016). Gctf: Real-timeCTF determination and correction. J Struct Biol 193, 1-12.
转载本文请联系原作者获取授权,同时请注明本文来自张凯科学网博客。
链接地址:http://blog.sciencenet.cn/blog-972184-1064793.html
 |
评分
-
查看全部评分
|










 发表于 2017-7-5 21:03:09
发表于 2017-7-5 21:03:09

 收藏
收藏 发表于 2018-5-17 02:29:59
发表于 2018-5-17 02:29:59
